Examples
Identification of conserved waters from molecular dynamics trajectories
The most freqeunt use case is one where a molecular dynamics (MD) trajectory is analysed for identification of conserved waters.
Preprocessing of trajectories using WaterNetworkAnalysis
The first step is to align the trajectory on to the desired snapshot. If this is done by user, one should make sure to save the snapshot onto which the trajectory was aligned to for visualisation of results.
Since clustering procedure does not employ periodic boundary conditions, the simulation box should be centered in such a way that the center of the region within which conserved water molecules are studied is in the centere of the simulation box. Alternatively, users should ensure that the region of interest does not span across periodic images and belongs to a single periodic image and does not cross the periodic boundary.
Next, water atom coordinates in specific simulation region should be extracted.The arrays containing this data must be set up in specific way. Each atom type in water molecule needs to be represented in a single numpy array or a list (3 in total - one for oxygen and two hydrogen arrays containing three dimensional coordiantes). Each row in each of these arrays has to belong to the same water molecule. Oxygen array should contain xyz coordinates, while hydrogen arrays should contain orientations from central oxygen atom instead. One can convert from an oxygen coordinates array and a hydrogen coordinates array using ConservedWaterSearch.utils.get_orientations_from_positions(). Hydrogen array should contain hydrogen coordinates of the same water molecule one after another in the same order as in oxygen coordinates array.
We host a seperate package which serves this purpose called WaterNetworkAnalysis (WNA). Since WNA uses MDAnalysis most file types can be read in for analysis. For more information on preprocessing trajectory data, please refer to the WaterNetworkAnalysis documentation.
Identification of conserved waters using only oxygen data
The central object for determination of conserved waters from MD trajectories is by using ConservedWaterSearch.water_clustering.WaterClustering class. In this example we shall perform Single Clustering (SC) procedure which is extremely fast, but produces incomplete results in most cases. This means that some conserved waters won’t be properly detected. In this example we shall perform clustering on oxygen atoms only which will produce even worse results but will be extremely fast.
# imports
import numpy as np
from ConservedWaterSearch.water_clustering import WaterClustering
from ConservedWaterSearch.utils import get_orientations_from_positions
# Number of snapshots
Nsnap = 20
# load some example - trajectory should be aligned prior to extraction of atom coordinates
Opos = np.loadtxt("tests/data/testdataO.dat")
wc = WaterClustering(nsnaps=Nsnap)
wc.single_clustering(Opos, [], [], whichH=['onlyO'])
# "aligned.pdb" should be the snapshot original trajectory was aligned to.
wc.visualise_pymol(aligned_protein = "aligned.pdb", output_file = "waters.pse")
Identification of conserved waters using full water data
In this example we shall perform Multi Stage Re-Clustering (MSRC) procedure which is slow, but produces excellent results in most cases. This means that most conserved waters will be properly detected. In this example we shall perform clustering on both oxygen and hydrogen coordination data.
# imports
import numpy as np
from ConservedWaterSearch.water_clustering import WaterClustering
from ConservedWaterSearch.utils import get_orientations_from_positions
# Number of snapshots
Nsnap = 20
# load some example - trajectory should be aligned prior to extraction of atom coordinates
Opos = np.loadtxt("tests/data/testdataO.dat")
Hpos = np.loadtxt("tests/data/testdataH.dat")
wc = WaterClustering(nsnaps=Nsnap)
wc.multi_stage_reclustering(*get_orientations_from_positions(Opos, Hpos))
print(wc.water_type)
# "aligned.pdb" should be the snapshot original trajectory was aligned to.
wc.visualise_pymol(aligned_protein = "aligned.pdb", output_file = "waters.pse")
Pymol session similar to the following one should be produced:
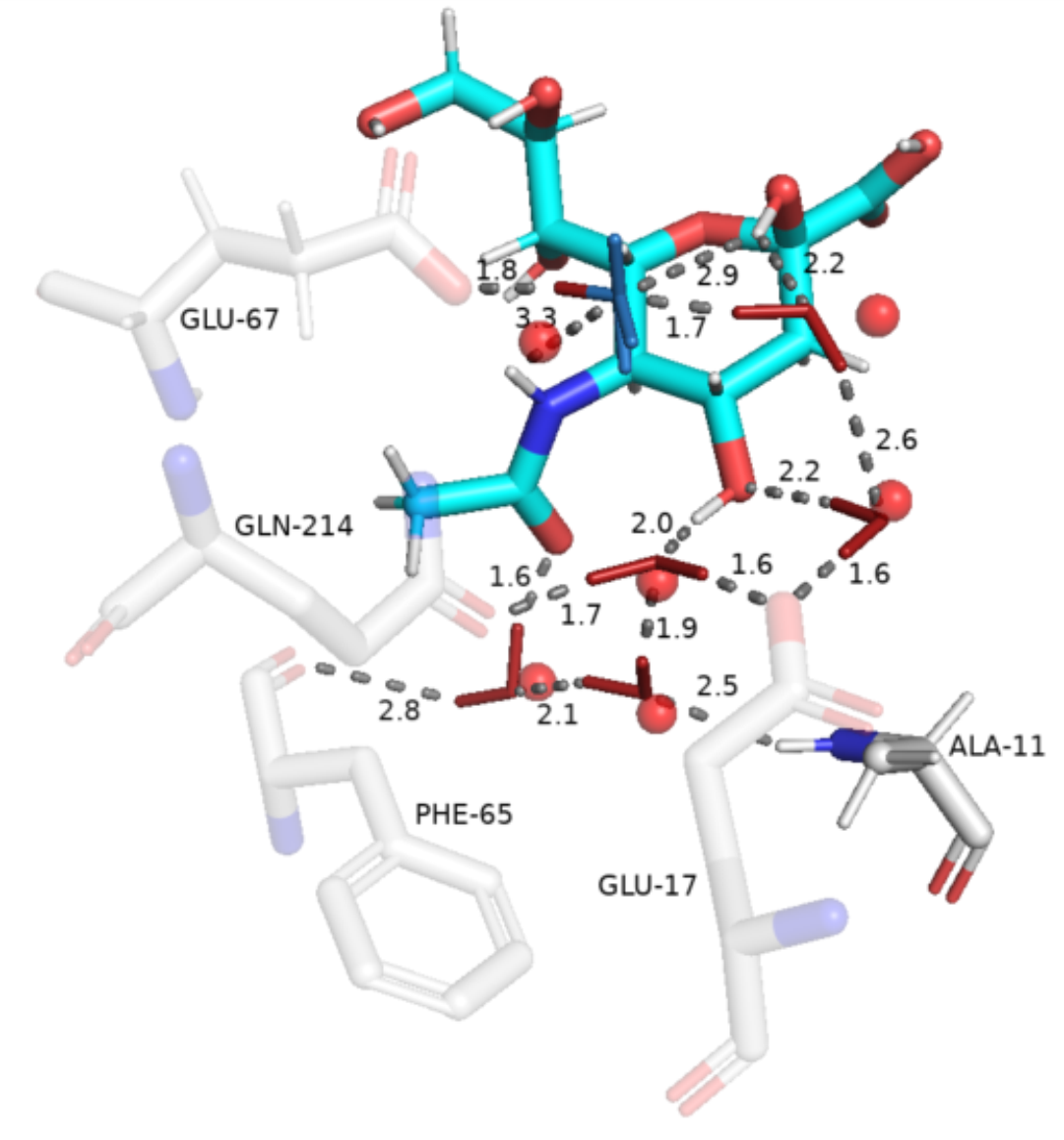
Determination of conserved water types from hydrogen orientation data
Given a set of hydrogen orientations, one can classify the water molecule to which the conserved water belongs to based on the proposed scheme:
import ConservedWaterSearch.hydrogen_orientation as HO
# load some example
orientations = np.loadtxt("tests/data/conserved_sample_FCW.dat")
# Run classification
res = HO.hydrogen_orientation_analysis(
orientations,
)
# print averaged hydrogen orientations and the water type
print(i for i in res)
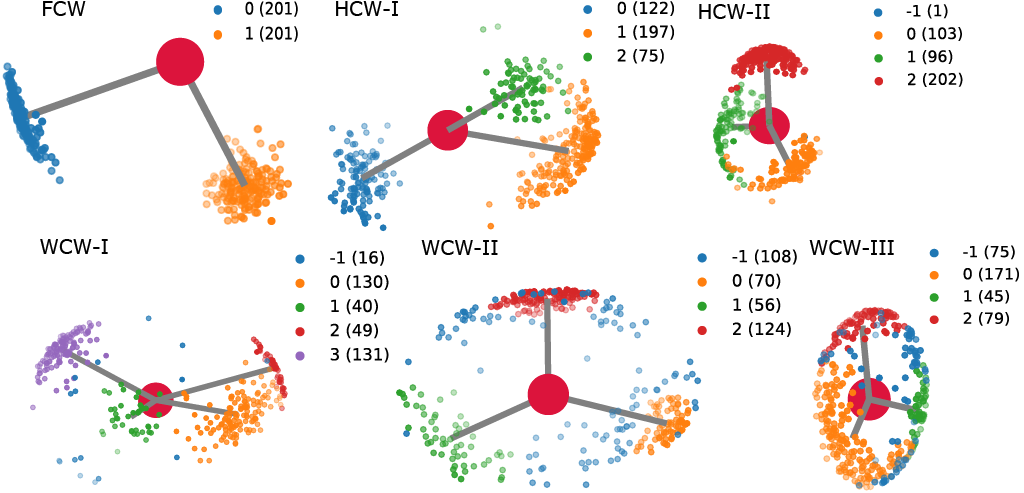
Note that for HCW and WCW multiple average hydrogen orientation pairs can be produced. For example in the figure above, HCW-I should produce such orientation pairs. In the first pair, hydrogen 1 will be oriented in the direction of cluster 1 (orange) and hydrogen 2 will be oriented in the direction of cluster 0 (blue). In the second pair, hydrogen 1 will again be oriented in the direction of cluster 1 (orange) and hydrogen 2 will be oriented in the direction of cluster 2 (green). This is because the main hydrogen is defined by the largest cluster which in this case is cluster 1 (orange). For more information see here.
Checking specific water types
In case the user is interested in only classifying for some conserved water types, the type specific functions can be used. However, prefered way to do this is to optional parameter to the ConservedWaterSearch.hydrogen_orientation.hydrogen_orientation_analysis():
import ConservedWaterSearch.hydrogen_orientation as HO
# load some example
orientations = np.loadtxt("tests/data/conserved_sample_FCW.dat")
# Run classification for FCW and HCW only
res = HO.hydrogen_orientation_analysis(
orientations,
which = ['FCW','HCW']
)
# print averaged hydrogen orientations and the water type
print(i for i in res)